
Microbial protein structure
Methane is an end product of the anaerobic degradation of organic materials. This combustible gas is useful as a fuel but also it is a potential greenhouse gas (1). Huge amounts of methane are produced by methanogenic archaea on Earth. Increasing anthropogenic methane production could become a global issue. We are interested in the enzymes involved in methanogenesis from H2 and CO2 (Figure 1). In the hydrogenotrophic methanogenic pathway, many unique enzymes are involved (2). These methanogenic enzymes contain novel cofactors and use unique coenzymes as the substrates. My group is mainly working on three major projects on the methanogenic enzymes:
- Structure and function of the enzymes involved in hydrogenotrophic methanogenesis.
- Catalytic mechanism of [Fe]-hydrogenase.
- Biosynthesis of the [Fe]-hydrogenase cofactor.
Structure and function of the enzymes involved in methanogenesis
![Figure 1. Methane formation from H2 and CO2 in hydrogenotrophic methanogens. 1. Formyl-methanofuran dehydrogenase (Fwd); 2. Formyl-methanofuran:H4MPT formyltransferase (Ftr); 3. Methenyl-H4MPT+ cyclohydrolase (Mch); 4a. H2-forming methylene-H4MPT dehydrogenase ([Fe]-hydrogenase or Hmd); 4b. F420-dependent methylene-H4MPT dehydrogenase (Mtd); 5. F420-reducing hydrogenase (Frh); 6. F420-dependent methylene-H4MPT reductase (Mer); 7. Membrane-spanning methyl-H4MPT:coenzyme M methyltransferase (MtrA–H); 8. Methyl-coenzyme M reductase (Mcr); 9. Heterodisulfide-reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD); 10, A1AO ATP synthase and 11, energy-converting [NiFe]-hydrogenases (Eha and Ehb).](/1022948/original-1613664316.webp?t=eyJ3aWR0aCI6OTYwLCJmaWxlX2V4dGVuc2lvbiI6IndlYnAiLCJvYmpfaWQiOjEwMjI5NDh9--f000ce4021d8d862b4acf8db060ab35248b571cd "Figure 1. Methane formation from H2 and CO2 in hydrogenotrophic methanogens. 1. Formyl-methanofuran dehydrogenase (Fwd); 2. Formyl-methanofuran:H4MPT formyltransferase (Ftr); 3. Methenyl-H4MPT+ cyclohydrolase (Mch); 4a. H2-forming methylene-H4MPT dehydrogenase ([Fe]-hydrogenase or Hmd); 4b. F420-dependent methylene-H4MPT dehydrogenase (Mtd); 5. F420-reducing hydrogenase (Frh); 6. F420-dependent methylene-H4MPT reductase (Mer); 7. Membrane-spanning methyl-H4MPT:coenzyme M methyltransferase (MtrA–H); 8. Methyl-coenzyme M reductase (Mcr); 9. Heterodisulfide-reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD); 10, A1AO ATP synthase and 11, energy-converting [NiFe]-hydrogenases (Eha and Ehb).")
![Figure 1. Methane formation from H2 and CO2 in hydrogenotrophic methanogens. 1. Formyl-methanofuran dehydrogenase (Fwd); 2. Formyl-methanofuran:H4MPT formyltransferase (Ftr); 3. Methenyl-H4MPT+ cyclohydrolase (Mch); 4a. H2-forming methylene-H4MPT dehydrogenase ([Fe]-hydrogenase or Hmd); 4b. F420-dependent methylene-H4MPT dehydrogenase (Mtd); 5. F420-reducing hydrogenase (Frh); 6. F420-dependent methylene-H4MPT reductase (Mer); 7. Membrane-spanning methyl-H4MPT:coenzyme M methyltransferase (MtrA–H); 8. Methyl-coenzyme M reductase (Mcr); 9. Heterodisulfide-reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD); 10, A1AO ATP synthase and 11, energy-converting [NiFe]-hydrogenases (Eha and Ehb).](/1022948/original-1613664316.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjEwMjI5NDh9--4b378347f2b924cb4385dac31afa672b13106e1f)
In this project, my group intensively collaborates with Ulrich Ermler (X-ray crystallography) from the Max Planck Institute of Biophysics in Frankfurt. We have elucidated the structure of formyl-methanofuran dehydrogenase (Fwd) (3), formyl-methanofuran:tetrahydromethanopterin (H4MPT) formyltransferase (4-6), methenyl-H4MPT+-cyclohydrolase (Mch) (7, 8), F420-dependent methylene-H4MPT dehydrogenase (Mtd) (9, 10), H2-forming methylene-H4MPT dehydrogenase ([Fe]-hydrogenase or Hmd) (11-14), methylene-H4MPT reductase (Mer) (15, 16), the MtrA subunit of membrane-spanning methyl-H4MPT:coenzyme M methyltransferase complex (MtrA–H) (17), methyl-coenzyme M reductase (Mcr) (18-22), Heterodisulfide-reductase/[NiFe]-hydrogenase complex (Hdr/Mvh) (23), F420-reducing [NiFe]-hydrogenase (Frh) (24-26), F420H2 oxidase (FprA) (27, 28), and F420-dependent alcohol dehydrogenase (29) from methanogenic archaea. In addition, we solved the structures of acetoacetyl-CoA thiolase/HMG-CoA synthase complex (30) and HMG-CoA reductase (31) from a methanogenic archaeon, F420:NADP oxidoreductase from a sulfate-reducing archaeon (32), NADP dependent methylene-H4MPT dehydrogenase (33) and formyltransferase-hydrolase complex (34) from a methylotrophic bacterium, and methyl-coenzyme M reductase from a methylotrophic archaeon (35, 36). As examples, the structure and function of Fwd and Mvh-Hdr are described below.
The formyl-methanofuran dehydrogenase complex is organized in a tetramer of heterohexamer built up with FwdABCDFG. (B)The catalytic module FwdABDG:CO2 enters the formate dehydrogenase center via a channel (blue mesh). Electrons from ferredoxin reduce the CO2 to formate. The formate molecule is transferred to the amidohydrolase Zn-Zn center via an inner cavity (red mesh). Formate reacts with methanofuran (MFR) to generate formyl-MFR (CHO-MFR).")

Figure 2. Structure and the reaction scheme of the Fwd complex. (A)The formyl-methanofuran dehydrogenase complex is organized in a tetramer of heterohexamer built up with FwdABCDFG. (B)The catalytic module FwdABDG:CO2 enters the formate dehydrogenase center via a channel (blue mesh). Electrons from ferredoxin reduce the CO2 to formate. The formate molecule is transferred to the amidohydrolase Zn-Zn center via an inner cavity (red mesh). Formate reacts with methanofuran (MFR) to generate formyl-MFR (CHO-MFR).
Formyl-methanofuran dehydrogenase (Fwd). Hydrogenotrophic methanogens start the metabolism by an ATP-independent CO2 fixation process. This reaction is catalyzed by formyl-methanofuran dehydrogenase (Fwd), which fixes CO2 on the methanofuran coenzyme to form formyl-methanofuran (Figure 1, Reaction 1). We solved the crystal structure of the Fwd complex (3). Fwd is organized in a tetramer of heterohexamer, Fwd(ABCDFG)4,which contains 46 [4Fe-4S] clusters (Figure 2A). Most biological CO2 fixation metabolisms start with trapping CO2 as a carboxy group, which is followed by a reduction. In contrast, Fwd uses a different strategy, where CO2 is firstly reduced to formate, which is then fixed on methanofuran to generate formyl-methanofuran (Figure 2B). The subunit FwdB, which is homologous to formate dehydrogenase, contains a tungstopterin active site and a channel for CO2 entry. Electrons are transferred through the [4Fe-4S] clusters to the tungstopterin for the CO2 reduction to formate. The formate formed will be sent to the next active site through a 43-Å-long tunnel. FwdA is a homolog of amidohydrolase and catalyzes the condensation of formate to methanofuran. One of the unexpected features of the 800-kDa complex is the electronic wire composed of 46 [4Fe-4S]-clusters (Figure 2A). The arrangement of [4Fe-4S] clusters functions as an electron relay, and may synchronize the four tungstopterin active sites over 206 Å.


Figure 3. The three-dimensional structure the HdrABC-MvhAGD complex and the catalyzed reactions.The arrows with solid- and broken-lines indicate the reactions and the electron pathways, respectively. One HdrABC-MvhAGD part is shown by cartoon model and transparent surface-model with colors and another HdrABC-MvhAGD part is shown by gray surface-model.
Heterodisulfide-reductase/[NiFe]-hydrogenase complex (Hdr/Mvh). The final step of the methanogenic pathway is catalyzed by Mcr (Figure 1, Reaction 8), which generates two products: methane and the heterodisulfide (CoM-S-S-CoB). The reduced coenzyme M and B are regenerated from CoM-S-S-CoB by the reaction catalyzed by heterodisulfide-reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD) (Figure 1, Reaction 9). HdrA is the catalytic center of a new mode of energy-coupling mechanism “flavin-based electron bifurcation”. In this process, 4 electrons are separated into 2 electrons with lower energy and 2 electrons with higher energy for reduction of heterodisulfide and ferredoxin, respectively. The reduced ferredoxin gives high-energy electrons to CO2, which is catalyzed by formyl-methanofuran dehydrogenases (Fwd) described above. To explore the catalytic mechanism of the HdrABC-MvhAGD complex, we solved the crystal structure of the HdrABC-MvhAGD complex from a methanogen Methanothermococcus thermolithotrophicus (23). The crystal structure allowed us to draw the plausible electron transfer pathways from the hydrogenase to the heterodisulfide reductase modules in this enzyme complex (Figure 3). Location of flavin and the conserved amino-acid residues at the flavin binding-site in HdrA supported the proposed function of flavin in the electron-bifurcation mechanism. HdrA homologs are found in various enzyme complexes in numerous microorganisms. The HdrA structure of the HdrABC-MvhAGD complex serves as the prototype of this protein family. In addition, in the HdrB subunit, we found a new type of iron-sulfur cluster, noncubane [4Fe-4S] cluster, which is bound to the amino-acid residues at the CCG motifs. The CCG motif is found in 2000 other proteins but the structure and function was unknown. Cryo-intermediate-trapping experiments indicated that the noncubane iron-sulfur clusters performed the reduction of the CoM-S-S-CoB.
Recently, we solved the structure of a 3-Mda complex of formate dehydrogenase (Fdh), Hdr, and formylmethanofuran dehydrogenase (Fmd) in a hexamer of the Fdh-Hdr-Fmd dimer from Methanospirillum hungatei (63). This megacomplex enzyme catalyzes the reduction of CO2 to formyl-MFR with low potential electrons provided by the flavin-based electron bifurcation mechanism in Hdr using electrons from formate and the reduced form of F420. The low-potential electron-output domain of Hdr and the CO2-reducing domain in Fmd are linked by electron chains of the FmdF polyferredoxin.
The catalytic mechanism of [Fe]-hydrogenase
![Figure 4. Open/closed conformational changes of [Fe]-hydrogenase triggered by binding of the substrate methenyl-H4MPT+. Upon the open-to-closed conformational change, the coordination of the iron site of the FeGP cofactor changed from 6 coordination to 5 coordination.](/1023357/original-1613665099.webp?t=eyJ3aWR0aCI6OTYwLCJmaWxlX2V4dGVuc2lvbiI6IndlYnAiLCJvYmpfaWQiOjEwMjMzNTd9--c8d9d45833cd4741fbdd42551c7d103124c799ad "Figure 4. Open/closed conformational changes of [Fe]-hydrogenase triggered by binding of the substrate methenyl-H4MPT+. Upon the open-to-closed conformational change, the coordination of the iron site of the FeGP cofactor changed from 6 coordination to 5 coordination.")
![Figure 4. Open/closed conformational changes of [Fe]-hydrogenase triggered by binding of the substrate methenyl-H4MPT+. Upon the open-to-closed conformational change, the coordination of the iron site of the FeGP cofactor changed from 6 coordination to 5 coordination.](/1023357/original-1613665099.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjEwMjMzNTd9--94f8245562df845f4ee533745c98ccd7edc0c6ac)
Figure 4. Open/closed conformational changes of [Fe]-hydrogenase triggered by binding of the substrate methenyl-H4MPT+. Upon the open-to-closed conformational change, the coordination of the iron site of the FeGP cofactor changed from 6 coordination to 5 coordination.
Molecular hydrogen and proton are used as electron donor or acceptor, respectively, in the metabolism of many anaerobic microorganisms. Hydrogenases catalyze the reversible reaction (37). The most prominent hydrogenases are [NiFe]-hydrogenase and [FeFe]-hydrogenase. [Fe]-hydrogenase (Hmd) is not related to either [NiFe]-hydrogenase or [FeFe]-hydrogenase (38). This third type of hydrogenases contains a mononuclear iron cofactor, iron-guanylylpyridinol (FeGP) cofactor cartooned in Figure 4 and 5. This enzyme catalyzes the reversible dehydrogenation of methylene-H4MPT to methenyl- H4MPT+. We solved the atomic-resolution (1.06 Å) structure of [Fe]-hydrogenase bound with substrate, methenyl-H4MPT+, in which the activated enzyme is ready for H2-binding and -activation at the mono-Fe site (11). Compared to the substrate-free [Fe]-hydrogenase, the closed active [Fe]-hydrogenase displays a different coordination structure of the Fe center. In the activated enzyme, the active-site cleft closed and the water ligand of the Fe site is dissociated from the Fe center, which results in the penta-coordinated Fe complex (Figure 4). The removal of the water ligand makes the open site accessible for the H2 binding. The structural change was supported by Mössbauer and infrared spectroscopic analyses. The deprotonated 2-OH group of the FegP cofactor acts as catalytic base, where H2-binding to Fe, H2-cleavage and finally hydride transfer proceeds smoothly at the active site. In the crystal structure, the substrate is highly distorted. Based on structural analyses and kinetic studies (13, 39-44), we illustrate a catalytic cycle of the [Fe]-hydrogenase (2, 11).
![Figure 5. The proposed catalytic cycle of [Fe]-hydrogenase.](/1023433/original-1613665099.webp?t=eyJ3aWR0aCI6OTYwLCJmaWxlX2V4dGVuc2lvbiI6IndlYnAiLCJvYmpfaWQiOjEwMjM0MzN9--f466ef2fa295be6e750fdab931b08a9ad85a024d "Figure 5. The proposed catalytic cycle of [Fe]-hydrogenase.")
![Figure 5. The proposed catalytic cycle of [Fe]-hydrogenase.](/1023433/original-1613665099.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjEwMjM0MzN9--4cf688e70c138fd4940c4113b274bd64d8f98b01)
Figure 5. The proposed catalytic cycle of [Fe]-hydrogenase.
We constructed active [Fe]-hydrogenase holoenzymes with a mimetic iron-pyridinol model compound and the apoenzyme in collaboration with the group of Xile Hu (Ecole polytechnique fédérale de Lausanne, EPFL) (45). The mimetic cofactor lacks the bulky GMP nucleotide and two methyl substituents compared to the native cofactor. Despite the modifications, the turnover frequency of the reconstituted enzyme was 1–2 s−1, which is much higher than that of synthetic hydrogenation catalysts. This is the first construction of an active metalloenzyme using a truncated metal cofactor. In addition, we showed that constructing the enzyme using a mimetic cofactor containing a 2-hydroxy pyridine group restores activity, while an analogous experiment with a 2-methoxy-pyridine complex leads to an inactive enzyme. These findings support a mechanism in which the 2-hydroxy group is deprotonated before serving as an internal base for heterolytic H2 cleavage. Our results open a door in the design of new mimetic active-site cofactors for hydrogenation and the production of H2, an important future energy currency. We have recently constructed a [Mn]-hydrogenase using a Mn model complex (46).
Biosynthesis of the [Fe]-hydrogenase cofactor

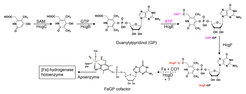
Figure 6. Proposed biosynthesis sequence of the FeGP cofactor biosynthesis.
Because of the unique structural and functional features of the FeGP cofactor (11, 14, 47-53), its biosynthesis is of great interest in chemistry and biology. Based on retro-synthetic analysis and stable-isotope-labeling data, we proposed a pathway for the FeGP cofactor biosynthesis involving various reactions for pyridinol formation, pyridinol methylation, and iron center formation (54, 55). In many methanogenic archaea, the hmd genes ([Fe]-hydrogenase structural genes) are clustered with hmd-co-occurring genes (hcgA–G), which suggests that the hcgA–G genes are involved in biosynthesis of the FeGP cofactor (37, 55). To analyze the function of the hcg genes, we employed a “structure–to-function” strategy.
HcgB. We first elucidated the function of the HcgB protein using this approach (56). Our comparative genomic studies revealed a structural relationship between HcgB and nucleotide triphosphatases, which was not ascertainable at the sequence level. Superimposed structures of HcgB and nucleotide triphosphatase indicated that the nucleotide triphosphate-binding cleft of nucleoside triphosphatase is similar to that of HcgB. However, a crucial aspartate, the base catalyst for hydrolysis of nucleoside triphosphates, is exchanged in HcgB with a glycine. Therefore, we predicted that HcgB has the ability to bind and activate nucleoside triphosphate, but lacks nucleoside triphosphatase activity. These findings sparked the idea that HcgB might catalyze a guanylyltransferase reaction to conjugate the pyridinol ring to GMP. To test this hypothesis, we used GTP and pyridinol model substrates in the presence of HcgB from Methanocaldococcus jannaschii in biosynthetic model reactions to form the corresponding guanylyl-pyridinol products. Using NMR, we identified 3,6-dimethyl-2,4-dihydroxypyridine-GMP as the model reaction product. In the cocrystal structure of HcgB from M. jannaschii guanylylpyridinol (GP) precursor was located inside the predicted substrate-binding cleft. Kinetic studies indicated that HcgB catalyzes formation of GP from the pyridinol precursors and GTP (55, 56).
HcgC. The pyridinol ring of the FeGP cofactor is substituted with two methyl groups, an acylmethyl group, and GMP. Crystal structure of HcgC from Methanocaldococcus jannaschii was solved and we found that HcgC was structurally similar to S-adenosylmethionine (SAM)-dependent methyltransferases, which suggested the potential function as a methyltransferase (57). Previous isotope-labeling experiment indicated that the 3-methyl group of the pyridinol ring was obtained from methionine most probably via SAM. The predicted precursor (methyl acceptor) of the HcgC reaction was chemically synthesized in collaboration with Xile Hu (EPFL). The methylation reaction catalyzed by HcgC was confirmed by experiments using the synthesized compound (57). Furthermore, we clarified the unique catalytic mechanism, which use water molecules to activate the pyridinol substrate (58).
HcgD. This enzyme is a homolog of the ubiquitous Nif3-like protein family proteins, which contain dinuclear metal center. HcgD contains a dinuclear iron center. One of the irons is removed by chelating reagents. This finding suggests that HcgD might have an iron-trafficking function in biosynthesis of the FeGP cofactor (59).
HcgE. The primary structure of HcgE resembles that of E1-like ubiquitin-activating enzymes (E1 enzymes). E1 enzymes catalyze adenylylation of the C-terminal end of ubiquitin(-like) proteins using ATP as adenylyl group donor. The structure of the co-crystal of HcgE with ATP and GP revealed that GP binds to the active site pocket of HcgE, where the carboxy group points towards the a-phosphate of ATP (60). This finding suggested that HcgE catalyzes adenylylation of the 6-carboxymethyl group of GP. The HcgE-catalyzed reaction was kinetically confirmed.
HcgF. This enzyme is not similar in primary structure to any proteins of known function. Co-crystal structures indicated that the HcgF homodimer has two GP-binding sites. One of two GP molecules is partially bound to HcgF by covalent bonding of the carboxyl group of GP to Cys9-thiolate (60). Such thioester bonding of the C-terminal carboxyl group and a cysteine-thiolate is observed in the ubiquitin-activation process. Our hypothesis based on chemical precedents is that the thioester reacts with an iron species to form the acyl and thiolate ligands to iron of the cofactor.
Based on the results, a biosynthetic sequence of the FeGP cofactor is proposed. A pyridinol precursor is synthesized from β-alanine (or aspartate) and 2,3-dihydroxy-4-oxo-pentanoate like compound, which were predicted by retrosynthesis. The 3 position of pyridinol is methylated and the 4 position is conjugated with GMP to form GP. The 6-carboxy group of GP pyridinol ring is activated by adenylylation reaction with ATP and activated via formation of a thioester bond in HcgF, which suggests Fe-acyl formation via a nucleophilic substitution reaction to allow the release of Cys9 of HcgF as thiolate.
HcgA. The function of HcgA was studied using an in vitro biosynthesis assay with cell extract from M. maripaludis mutants containing a reaction cooktail. The first known pyridinol precursor of FeGP cofactor is biosynthesized by the reaction catalyzed by HcgA (64, 65). HcgA is a radical SAM enzyme and forms the pyridinol precursor from an unknown precursor in the cell extract of M. maripaludis.
HcgG. The in vitro biosynthetic assays indicated that HcgG catalyzes the reactions involved in completing the iron complex structure of FeGP cofactor; biosynthesis of CO and acyl ligands and insertion of Fe (64, 65). The radical SAM reaction is involved in the HcgG catalyzed reaction.
References
1. S. Shima, G. Huang, T. Wagner, U. Ermler, Structural basis of hydrogenotrophic methanogenesis. Annu. Rev. Biochem. 74, 713-733 (2020).
2. G. F. Huang, T. Wagner, U. Ermler, S. Shima, Methanogenesis involves direct hydride transfer from H2 to an organic substrate. Nat. Rev. Chem. 4, 213-221 (2020).
3. T. Wagner, U. Ermler, S. Shima, The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 354, 114-117 (2016).
4. U. Ermler, M. C. Merckel, R. K. Thauer, S. Shima, Formylmethanofuran: tetrahydromethanopterin formyltransferase from Methanopyrus kandleri - new insights into salt-dependence and thermostability. Structure 5, 635-646 (1997).
5. B. Mamat et al., Crystal structures and enzymatic properties of three formyltransferases from archaea: environmental adaptation and evolutionary relationship. Protein Sci. 11, 2168-2178 (2002).
6. P. Acharya, E. Warkentin, U. Ermler, R. K. Thauer, S. Shima, The structure of formylmethanofuran:tetrahydromethanopterin formyltransferase in complex with its coenzymes. J. Mol. Biol. 357, 870-879 (2006).
7. V. Upadhyay et al., Structure and catalytic mechanism of N5,N10-methenyltetrahydromethanopterin cyclohydrolase. Biochemistry 51, 8435-8443 (2012).
8. W. Grabarse et al., The crystal structure of methenyltetrahydromethanopterin cyclohydrolase from the hyperthermophilic archaeon Methanopyrus kandleri. Struct. Fold. Des. 7, 1257-1268 (1999).
9. K. Ceh et al., Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry 48, 10098-10105 (2009).
10. C. H. Hagemeier et al., Coenzyme F420 dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) from Methanopyrus kandleri: A methanogenic enzyme with an unusual quarternary structure. J. Mol. Biol. 332, 1047-1057 (2003).
11. G. F. Huang et al., The atomic-resolution crystal structure of activated [Fe]-hydrogenase. Nat. Catal. 2, 537-543 (2019).
12. T. Hiromoto et al., The crystal structure of C176A mutated [Fe]-hydrogenase suggests an acyl-iron ligation in the active site iron complex. FEBS Lett. 583, 585-590 (2009).
13. T. Hiromoto, E. Warkentin, J. Moll, U. Ermler, S. Shima, The crystal structure of an [Fe]-hydrogenase-substrate complex reveals the framework for H2 activation. Angew. Chem. Int. Ed. 48, 6457-6460 (2009).
14. S. Shima et al., The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 321, 572-575 (2008).
15. S. W. Aufhammer et al., Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: Architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci. 14, 1840-1849 (2005).
16. S. Shima et al., Structure of coenzyme F420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J. Mol. Biol. 300, 935-950 (2000).
17. T. Wagner, U. Ermler, S. Shima, MtrA of the sodium ion pumping methyltransferase binds cobalamin in a unique mode. Sci. Rep. 6, (2016).
18. U. Ermler, W. Grabarse, S. Shima, M. Goubeaud, R. K. Thauer, Crystal structure of methyl coenzyme M reductase: The key enzyme of biological methane formation. Science 278, 1457-1462 (1997).
19. W. Grabarse et al., On the mechanism of biological methane formation: Structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J. Mol. Biol. 309, 315-330 (2001).
20. W. G. Grabarse, F. Mahlert, S. Shima, R. K. Thauer, U. Ermler, Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: Unusual amino acid modification, conservation and adaptation. J. Mol. Biol. 303, 329-344 (2000).
21. T. Wagner, J. Kahnt, U. Ermler, S. Shima, Didehydroaspartate modification in methyl-coenzyme M reductase catalyzing methane formation. Angew. Chem. Int. Ed. 55, 10630-10633 (2016).
22. T. Wagner, C.-E. Wegner, J. Kahnt, U. Ermler, S. Shima, Phylogenetic and structural comparisons of the three types of methyl-coenzyme M reductase from Methanococcales and Methanobacteriales. J. Bacteriol. 199, e00197-00117 (2017).
23. T. Wagner, J. Koch, U. Ermler, S. Shima, Methanogenic heterodisulfide reductase (HdrABC-MvhAGD) uses two noncubane [4Fe-4S] clusters for reduction. Science 357, 699-703 (2017).
24. S. Vitt et al., The F420-Reducing [NiFe]-hydrogenase complex from Methanothermobacter marburgensis, the first X-ray structure of a group 3 family member. J. Mol. Biol. 426, 2813-2826 (2014).
25. Y. Ilina et al., X-ray crystallography and vibrational spectroscopy reveal the key determinants of biocatalytic dihydrogen cycling by [NiFe] hydrogenases. Angew. Chem. Int. Ed. 58, 18710-18714 (2019).
26. D. J. Mills, S. Vitt, M. Strauss, S. Shima, J. Vonck, De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. eLife 2, (2013).
27. S. Engilberge, T. Wagner, P. Carpentier, E. Girard, S. Shima, Krypton-derivatization highlights O2-channeling in a four-electron reducing oxidase. Chem. Commun. 56, 10863-10866 (2020).
28. H. Seedorf et al., Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic Archaea catalyzing the reduction of O2 to H2O. FEBS J. 274, 1588-1599 (2007).
29. S. W. Aufhammer et al., Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure 12, 361-370 (2004).
30. B. Vogeli et al., Archaeal acetoacetyl-CoA thiolase/HMG-CoA synthase complex channels the intermediate via a fused CoA-binding site. Proc. Natl. Acad. Sci. USA 115, 3380-3385 (2018).
31. B. Vogeli, S. Shima, T. J. Erb, T. Wagner, Crystal structure of archaeal HMG-CoA reductase: insights into structural changes of the C-terminal helix of the class-I enzyme. FEBS Lett. 593, 543-553 (2019).
32. E. Warkentin et al., Structures of F420H2 : NADP(+) oxidoreductase with and without its substrates bound. EMBO J. 20, 6561-6569 (2001).
33. G. F. Huang et al., The hydride transfer process in NADP-dependent methylene-tetrahydromethanopterin dehydrogenase. J. Mol. Biol. 432, 2042-2054 (2020).
34. J. L. Hemmann, T. Wagner, S. Shima, J. A. Vorholt, Methylofuran is a prosthetic group of the formyltransferase/hydrolase complex and shuttles one-carbon units between two active sites. Proc. Natl Acad. Sci. USA 116, 25583-25590 (2019).
35. M. Krüger et al., A conspicuous nickel protein in microbial mats that oxidize methane anaerobically. Nature 426, 878-881 (2003).
36. S. Shima et al., Structure of a methyl-coenzyme M reductase from Black Sea mats that oxidize methane anaerobically. Nature 481, 98-101 (2012).
37. R. K. Thauer et al., Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu. Rev. Biochem. 79, 507-536 (2010).
38. S. Shima, U. Ermler, Structure and function of [Fe]-hydrogenase and its iron-guanylylpyridinol (FeGP) cofactor. Eur. J. Inorg. Chem., 963-972 (2011).
39. S. Shima, K. Ataka, Isocyanides inhibit [Fe]-hydrogenase with very high affinity. FEBS Lett 585, 353-356 (2011).
40. H. Tamura et al., Crystal structures of [Fe]-hydrogenase in complex with inhibitory isocyanides: implications for the H2 activation site. Angew. Chem. Int. Ed. 52, 9656-9659 (2013).
41. S. Vogt, E. J. Lyon, S. Shima, R. K. Thauer, The exchange activities of [Fe] hydrogenase (iron-sulfur-cluster-free hydrogenase) from methanogenic archaea in comparison with the exchange activities of [FeFe] and [NiFe] hydrogenases. J. Biol. Inorg. Chem. 13, 97-106 (2008).
42. S. Shima, S. Vogt, A. Gobels, E. Bill, Iron-chromophore circular dichroism of [Fe]-hydrogenase: the conformational change required for H2 activation. Angew. Chem. Int. Ed. 49, 9917-9921 (2010).
43. T. Wagner, G. F. Huang, U. Ermler, S. Shima, How [Fe]-hydrogenase from Methanothermobacter is protected against light and oxidative stress. Angew. Chem. Int. Ed. 57, 15056-15059 (2018).
44. G. F. Huang et al., Dioxygen sensitivity of [Fe]-hydrogenase in the presence of reducing substrates. Angew. Chem. Int. Ed. 57, 4917-4920 (2018).
45. S. Shima et al., Reconstitution of [Fe]-hydrogenase using model complexes. Nat. Chem. 7, 995-1002 (2015).
46. H. J. Pan et al., A catalytically active [Mn]-hydrogenase incorporating a non-native metal cofactor. Nat. Chem. 11, 669-675 (2019).
47. M. Korbas et al., The iron-sulfur cluster-free hydrogenase (Hmd) is a metalloenzyme with a novel iron binding motif. J. Biol. Chem. 281, 30804-30813 (2006).
48. E. J. Lyon et al., Carbon monoxide as an intrinsic ligand to iron in the active site of the iron-sulfur-cluster-free hydrogenase H2-forming methylenetetrahydromethanopterin dehydrogenase as revealed by infrared spectroscopy. J. Am. Chem. Soc. 126, 14239-14248 (2004).
49. E. J. Lyon et al., UV-A/blue-light inactivation of the 'metal-free' hydrogenase (Hmd) from methanogenic archaea - The enzyme contains functional iron after all. Eur. J. Biochem. 271, 195-204 (2004).
50. S. Shima et al., The cofactor of the iron-sulfur cluster free hydrogenase Hmd: Structure of the light-inactivation product. Angew. Chem. Int. Ed. 43, 2547-2551 (2004).
51. S. Shima, E. J. Lyon, R. K. Thauer, B. Mienert, E. Bill, Mössbauer studies of the iron-sulfur cluster-free hydrogenase: The electronic state of the mononuclear Fe active site. J. Am. Chem. Soc. 127, 10430-10435 (2005).
52. S. Shima et al., Evidence for acyl-iron ligation in the active site of [Fe]-hydrogenase provided by mass spectrometry and infrared spectroscopy. Dalton Trans. 41, 767-771 (2012).
53. S. Shima, M. Schick, H. Tamura, Preparation of [Fe]-hydrogenase from methanogenic archaea. Methods Enzymol. 494, 119-137 (2011).
54. M. Schick et al., Biosynthesis of the iron-guanylylpyridinol cofactor of [Fe]-hydrogenase in methanogenic archaea as elucidated by stable-isotope labeling. J. Am. Chem. Soc. 134, 3271-3280 (2012).
55. L. Bai et al., Towards artificial methanogenesis: biosynthesis of the [Fe]-hydrogenase cofactor and characterization of the semi-synthetic hydrogenase. Faraday Discuss. in press., (2017).
56. T. Fujishiro et al., Identification of the HcgB enzyme in [Fe]-hydrogenase-cofactor biosynthesis. Angew. Chem. Int. Ed. 52, 12555-12558 (2013).
57. T. Fujishiro et al., Identification of HcgC as a SAM-dependent pyridinol methyltransferase in [Fe]-hydrogenase cofactor biosynthesis. Angew. Chem. Int. Ed. 55, 9648-9651 (2016).
58. L. Bai et al., A water-bridged H-bonding network contributes to the catalysis of the SAM-dependent C-methyltransferase HcgC. Angew. Chem. Int. Ed. 56, 10806-10809 (2017).
59. T. Fujishiro, U. Ermler, S. Shima, A possible iron delivery function of the dinuclear iron center of HcgD in [Fe]-hydrogenase cofactor biosynthesis. FEBS Lett 588, 2789-2793 (2014).
60. T. Fujishiro, J. Kahnt, U. Ermler, S. Shima, Protein-pyridinol thioester precursor for biosynthesis of the organometallic acyl-iron ligand in [Fe]-hydrogenase cofactor. Nat. Commun. 6, (2015).
61. T. Fujishiro, K. Ataka, U. Ermler, S. Shima, Towards a functional identification of catalytically inactive [Fe]-hydrogenase paralogs. FEBS J. 282, 3412-3423 (2015).
62. T. Watanabe et al., The bacterial [Fe]-hydrogenase paralog hmdII uses tetrahydrofolate derivatives as substrates. Angew. Chem. Int. Ed. 58, 3506-3510 (2019).
63. Watanabe, T. Pfeil-Gardiner, O. Kahnt, J. Koch, J. Shima, S. Murphy, B.J. Three-megadalton complex of methanogenic electron-bifurcating and CO2-fixing enzymes. Science 373, 1151 - 1155 (2021).
64. Schaupp, S. Arriaza Gallardo, F. J. Pan, H. J. Kahnt, J. Angelidou, G. Paczia, N. Costa, K. Hu, X. Shima, S. In vitro biosynthesis of the [Fe]-hydrogenase cofactor verifies the proposed biosynthetic precursors. Angew. Chem. Int. Ed. 61, e202200994 (2022).
65. Arriaza-Gallardo, F. J. Schaupp, S. Zheng, Y.-C. Abdul-Halim, M. F. Hui-Jie, P. Kahnt, J. Angelidou, G. Paczia, N. Hu, X. Costa, K. Shima, S. The function of two radical-SAM enzymes, HcgA and HcgG, in the biosynthesis of the [Fe]-hydrogenase cofactor. Angew. Chem. Int. Ed. 61, e202213239 (2022).



![Reconstruction of a central [Fe]-Hydrogenase metallocofactor in a test tube](/1207025/original-1650443086.webp?t=eyJ3aWR0aCI6OTYwLCJoZWlnaHQiOjY0MCwiZml0IjoiY3JvcCIsImZpbGVfZXh0ZW5zaW9uIjoid2VicCIsIm9ial9pZCI6MTIwNzAyNX0%3D--4a07bdf43949e4bbb6196beb2cfa91eca0661dd1)


![<p>Atomic crystal structure of activated [Fe]-hydrogenase</p>](/636742/teaser-1716364267.webp?t=eyJ3aWR0aCI6OTYwLCJoZWlnaHQiOjY0MCwiZml0IjoiY3JvcCIsImZpbGVfZXh0ZW5zaW9uIjoid2VicCIsIm9ial9pZCI6NjM2NzQyfQ%3D%3D--bf6b2103b1c4ea5135e584ba374b9d53ff2a2222)
![<p>[Mn]-hydrogenase: A new step towards redesigning hydrogenases</p>](/584881/teaser-1560940570.webp?t=eyJ3aWR0aCI6OTYwLCJoZWlnaHQiOjY0MCwiZml0IjoiY3JvcCIsImZpbGVfZXh0ZW5zaW9uIjoid2VicCIsIm9ial9pZCI6NTg0ODgxfQ%3D%3D--0319b71b56ddde647e7a2e7b517552dbd490a760)
![<p>Discovery of [Fe]-hydrogenase in bacteria opens new possibilities for conversion of hydrogen</p>](/506336/original-1555497672.webp?t=eyJ3aWR0aCI6OTYwLCJoZWlnaHQiOjY0MCwiZml0IjoiY3JvcCIsImZpbGVfZXh0ZW5zaW9uIjoid2VicCIsIm9ial9pZCI6NTA2MzM2fQ%3D%3D--e5ec05ed8c1f23572f17f0422e4b79a402ee9ba7)
![Protection of [Fe]-hydrogenase](/416905/teaser-1561027823.webp?t=eyJ3aWR0aCI6OTYwLCJoZWlnaHQiOjY0MCwiZml0IjoiY3JvcCIsImZpbGVfZXh0ZW5zaW9uIjoid2VicCIsIm9ial9pZCI6NDE2OTA1fQ%3D%3D--c35a08047d48bfc25e0598260fec42fd23625a50)

