Mechanisms of Self-organisation within Bacterial Cells
Plasmid partitioning driven by collective migration of ParA between nucleoid lobes


The ParABS system is crucial for the faithful segregation and inheritance of many bacterial chromosomes and low-copy number plasmids. However, despite extensive research, the spatio-temporal dynamics of the ATPase ParA and its connection to the dynamics and positioning of the ParB-coated cargo has remained unclear. In this study, we utilise high-throughput imaging, quantitative data analysis, and computational modelling to explore the in vivo dynamics of ParA and its interaction with ParB-coated plasmids and the nucleoid. As previously observed, we find that F-plasmid ParA undergoes collective migrations (‘flips’) between cell halves multiple times per cell cycle. We reveal that a constricting nucleoid is required for these migrations and that they are triggered by a plasmid crossing into the cell half with greater ParA. Using simulations, we show that these dynamics can be explained by the combination of nucleoid constriction and cooperative ParA binding to the DNA, in line with the behaviour of other ParA proteins. We further show that these ParA flips act to equally partition plasmids between the two lobes of the constricted nucleoid and are therefore important for plasmid stability, especially in fast growth conditions for which the nucleoid constricts early in the cell cycle. Overall our work identifies a second mode of action of the ParABS system and deepens our understanding of how this important segregation system functions.
Robin Köhler and Seán M. Murray† (2024), PNAS 121(18) e2319205121, https://www.pnas.org/doi/10.1073/pnas.2319205121
Mid-cell migration of the chromosomal terminus is coupled to origin segregation in Escherichia coli


Bacterial chromosomes are dynamically and spatially organised within cells. In slow-growing Escherichia coli, the chromosomal terminus is initially located at the new pole and must therefore migrate to midcell during replication to reproduce the same pattern in the daughter cells. Here, we use high-throughput time-lapse microscopy to quantify this transition, its timing and its relationship to chromosome segregation. We find that terminus centralisation is a rapid discrete event that occurs ~25 min after initial separation of duplicated origins and ~50 min before the onset of bulk nucleoid segregation but with substantial variation between cells. Despite this variation, its movement is tightly coincident with the completion of origin segregation, even in the absence of its linkage to the divisome, suggesting a coupling between these two events. Indeed, we find that terminus centralisation does not occur if origin segregation away from mid-cell is disrupted, which results in daughter cells having an inverted chromosome organisation. Overall, our study quantifies the choreography of origin-terminus positioning and identifies an unexplored connection between these loci, furthering our understanding of chromosome segregation in this bacterium.
Ismath Sadhir and Seán M. Murray† (2023), Nature Communications 14 (1), 7489 https://dx.doi.org/10.1038/s41467-023-43351-7
Inferred counting and tracking of replicating DNA loci


Fluorescent microscopy is the primary method to study DNA organization within cells. However the variability and low signal-to-noise commonly associated with live-cell time lapse imaging challenges quantitative measurements. In particular, obtaining quantitative or mechanistic insight often depends on the accurate tracking of fluorescent particles. Here, we present ★Track, an inference method that determines the most likely temporal tracking of replicating intracellular particles such DNA loci while accounting for missing, merged and spurious detections. It allows the accurate prediction of particle copy numbers as well as the timing of replication events. We demonstrate ★Track’s abilities and gain new insight into plasmid copy number control and the volume dependence of bacterial chromosome replication initiation. By enabling the accurate tracking of DNA loci, ★Track can help to uncover the mechanistic principles of chromosome organisation and dynamics across a range of systems.
Robin Köhler, Ismath Sadhir and Seán M. Murray† (2023), Biophysical Journal 122, 1577-1585 (Cover image, May 2 issue; Selected as one of the best 5 Biophysical Journal articles of 2023) https://doi.org/10.1016/j.bpj.2023.03.033
Organisation of the ParABS partition complex


Chromosome segregation is vital for cell replication and in many bacteria is controlled by the ParABS system. A key part of this machinery is the association of ParB proteins to the parS-containing centromeric region to form the partition complex. Despite much work, the formation and structure of this nucleoprotein complex has remained unclear. However, it was recently discovered that CTP binding allows ParB dimers to entrap and slide along the DNA, as well as leading to more efficient condensation through ParB-mediated DNA bridging. Here, we use semi-flexible polymer simulations to show how these properties of sliding and bridging can explain partition complex formation. We find that transient ParB bridges can organise the DNA into either a globular state or into hairpins and helical structures, depending on the bridge lifetime. In separate stochastic simulations, we show that ParB sliding reproduces the experimentally measured multi-peaked binding profile of \textit{Caulobacter crescentus}, indicating that bridging and other potential roadblocks are sufficiently short-lived that they do not hinder ParB spreading. Indeed, upon coupling the two simulation frameworks into a unified sliding and bridging model, we find that short-lived ParB bridges do not hinder ParB sliding and the model can reproduce both the ParB binding profile and the condensation of the nucleoprotein complex. Overall, our model clarifies the mechanism of partition complex formation and predicts its fine structure. We speculate that the DNA hairpins produced by ParB bridging underlie the secondary function of ParB to load the Structural Maintenance of Chromosome (SMC) complex onto the DNA.
Lara Connolley, Lucas Schnabel, Martin Thanbichler and Seán M. Murray† (2023), Nature Communications, 14:4567, https://doi.org/10.1038/s41467-023-40320-y
Chromosome dynamics


Chromosomal loci in bacterial cells show a robust sub-diffusive scaling of the mean square displacement, MSD(\tau) \sim \tau^{\alpha}, with \alpha < 0.54. On the other hand, recent experiments have also shown that DNA-bridging Nucleoid Associated Proteins (NAPs) play an important role in chromosome organisation and compaction. Here, using polymer simulations we investigate the role of DNA bridging in determining the dynamics of chromosomal loci. We find that bridging compacts the polymer and reproduces the sub-diffusive elastic dynamics of monomers at timescales shorter than the bridge lifetime. Consistent with this prediction, we measure a higher exponent in a strain lacking the NAP H-NS compared to wild-type E. coli. Furthermore, bridging can reproduce the rare but ubiquitous rapid movements of chromosomal loci that have been observed in experiments. In our model the scaling exponent defines a relationship between the abundance of bridges and their lifetime. Using this and the observed mobility of chromosomal loci, we predict a lower bound on the average bridge lifetime of around 5 seconds.
Srikanth Subramanian and Seán M. Murray† (2023) Physical Review Research 5, 023034, https://doi.org/10.1103/PhysRevResearch.5.023034
Plasmid positioning by ParABS


The faithful segregation and inheritance of bacterial chromosomes and low-copy number plasmids requires dedicated partitioning systems. The most common of these, ParABS, consists of ParA, a DNA-binding ATPase and ParB, a protein that binds to centromeric-like parS sequences on the DNA cargo. The resulting nucleoprotein complexes are believed to move up a self-generated gradient of nucleoid-associated ParA. However, it remains unclear how this leads to the observed cargo positioning and dynamics. In particular, the evaluation of models of plasmid positioning has been hindered by the lack of quantitative measurements of plasmid dynamics. In this work, we used high-throughput imaging, analysis and modelling to determine the dynamical nature of these systems. We found that F plasmid is actively brought to specific subcellular home positions within the cell with dynamics akin to an over-damped spring and developed a unified stochastic model that quantitatively explains this behaviour and predicts that cells with the lowest plasmid concentration transition to oscillatory dynamics. We confirmed this prediction for F plasmid as well as a distantly-related ParABS system. Our results indicate that ParABS regularly positions plasmids across the nucleoid but operates just below the threshold of an oscillatory instability, which according to our model, minimises ATP consumption. This work also clarifies how various plasmid dynamics are achievable in a single unified stochastic model.
Robin Köhler, Eugen Kaganovitch and Seán M. Murray† (2022) eLife 11:e78743, https://doi.org/10.7554/eLife.78743
Seán M. Murray†, Martin Howard† (2019) Journal of Molecular Biology, https://doi.org/10.1016/j.jmb.2019.01.017, [arXiv: 1901.06139]
Tol-Pal relocalisation in Escherichia coli
 Major components of the Tol system. The lipoprotein Pal non-covalently associates with the cell wall. Pal also binds to TolB through an interaction that is mutually exclusive of peptidoglycan binding. TolA spans the periplasm and is coupled to the PMF through interactions with inner membrane components. The C-terminal domain of TolA interacts with the N-terminus of TolB. (b) Top, Average kymograph of Pal-mCherry during FRAP experiments of dividing wild-type cells. Colour indicates normalised fluorescence. Bottom, simulated kymograph obtained by fitting to the data above. (c) The mean Deff of Pal-mCherry as a function of cellular location in non-dividing and dividing cells obtained from FRAP data by fitting as in (b). Pal diffusion increases in dividing cells except at the septum where its mobility is similar to that in non-dividing cells.")
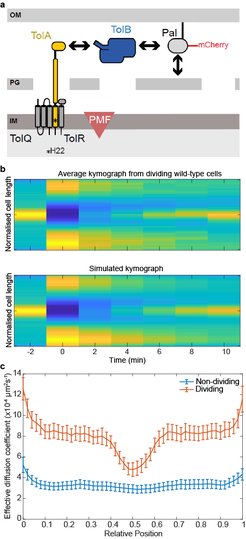
(a) Major components of the Tol system. The lipoprotein Pal non-covalently associates with the cell wall. Pal also binds to TolB through an interaction that is mutually exclusive of peptidoglycan binding. TolA spans the periplasm and is coupled to the PMF through interactions with inner membrane components. The C-terminal domain of TolA interacts with the N-terminus of TolB. (b) Top, Average kymograph of Pal-mCherry during FRAP experiments of dividing wild-type cells. Colour indicates normalised fluorescence. Bottom, simulated kymograph obtained by fitting to the data above. (c) The mean Deff of Pal-mCherry as a function of cellular location in non-dividing and dividing cells obtained from FRAP data by fitting as in (b). Pal diffusion increases in dividing cells except at the septum where its mobility is similar to that in non-dividing cells.
Gram-negative bacteria are protected from environmental insult and antibiotic attack by an additional protective layer, the outer membrane. During cell division, this membrane must be constricted at the septum in coordination with the invaginating cell wall. Recent studies have suggested this coordination is through the accumulation at the division site of Pal, an abundant lipoprotein that connects the outer membrane directly to the peptidoglycan of the cell wall. While the Tol transporter system is believed to be responsible for this reorganisation, the mechanism is unknown. In collaboration with Colin Kleanthous (Oxford), we have recently proposed how the Tol system can actively redistribute Pal within a dividing cell (Szczepaniak et al., 2019). We found that binding to TolB enhances the mobility of Pal molecules by preventing them binding to peptidoglycan. At the division septum, the inner membrane protein TolA, physically removes TolB from these complexes, thereby releasing Pal directly at the septum. This process is dependent on the proton-motive force in a similar manner to the ubiquitous TonB-dependent transporters. Our findings indicate that a diffusion-and-capture mechanism mediated by the PMF underlies the dynamic redistribution of immobile Pal molecules toward the division site, thereby coordinating outer membrane constriction with cell division.
As part of this work, we developed a new method for analysing Fluorescence Recovery After Photobleaching (FRAP) experiments, which we call SpatialFRAP. We used the Fokker-Planck formulism of diffusion in inhomogeneous media to obtain a spatially-varying effective diffusion constant across the long-axis of the cell. Using this, we discovered that the accumulation of Pal at the septum of dividing cells is not due to slower Pal mobility at that location but rather faster mobility everywhere else. This formed the basis for our proposed model. Since it uses information from across the entire cell, rather than just a specific region of interest, this method can measure very slowing diffusing processes within a reasonable short experiment time. For example, using 10 min FRAP experiments, we found that Pal diffuses with a very slow effective diffusion constant of ~3x10-4 µm2s-1. This would not be possible with the standard approach and is much lower than can be measured by single particle tracking. More recently, we used these results to develop and tested a mathematical model of how Tol redistributes slowly diffusing Pal molecules to the division septum (Connolley et al., 2021).
Connolley, Szczepaniak, Kleanthous† and Murray† (2021) PLoS Computational Biology 17(12): e1009756, https://doi.org/10.1371/journal.pcbi.1009756
Szczepaniak et al. (2020) Nature Communications, 11, 1305, https://doi.org/10.1038/s41467-020-15083-5
The role of MukBEF in the spatial positioning of ori
 A strain with FROS (LacI-mCherry) labelled ori and MukB-YPet was treated with DL serine hydroxamate to obtain cells with a single non-replicating chromosome and imaged at 1 min intervals. (b) The step-wise velocity of ori as a function of position relative to mid-cell (blue) and to the MukB focus (red). Shaded regions indicate standard error. Ori display a stronger attraction towards MukBEF foci than toward mid-cell. (c) A kymograph of simulated ori positioning (white lines) in the presence of self-organising MukBEF (colour-scale) in a growing cell. Correct partitioning is achieved if MukBEF is preferentially loaded onto the chromosome at ori and if ori experience a short-range repulsion (due for example to entropic repulsion of DNA polymers).")
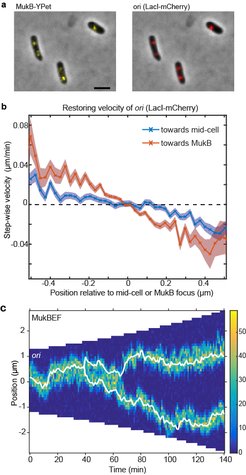
(a) A strain with FROS (LacI-mCherry) labelled ori and MukB-YPet was treated with DL serine hydroxamate to obtain cells with a single non-replicating chromosome and imaged at 1 min intervals. (b) The step-wise velocity of ori as a function of position relative to mid-cell (blue) and to the MukB focus (red). Shaded regions indicate standard error. Ori display a stronger attraction towards MukBEF foci than toward mid-cell. (c) A kymograph of simulated ori positioning (white lines) in the presence of self-organising MukBEF (colour-scale) in a growing cell. Correct partitioning is achieved if MukBEF is preferentially loaded onto the chromosome at ori and if ori experience a short-range repulsion (due for example to entropic repulsion of DNA polymers).
The starting point for chromosomal replication, the ori, has been found to have a crucial role in chromosome organisation and segregation. Not only is it duplicated and segregated first but its genomic position defines, in part, the boundaries of large interaction domains and its spatial position may determine the overall spatial organisation of the chromosome. In slowly to moderately growing Escherichia coli cells, the ori is first positioned at mid-cell. Following replication, duplicated ori separate and migrate, initially very rapidly, to opposite quarter positions, which become the new home positions for the remainder of the cell cycle. The mechanism underlying this behaviour has long been a mystery.
However, there is increasing evidence that MukBEF plays a role in origin positioning. MukBEF clusters (visualised as fluorescent foci) are found in close association with ori, at the middle or quarter positions and the splitting and movement of these foci occurs concurrently with the segregation of ori to the quarter positions. Depletion of MukF results in ori mis-positioning, which is restored upon repletion, suggesting a connection between their positioning. On the other hand, depletion of the type II topoisomerase Topo IV results in catenation and mis-positioning of ori but not of MukBEF foci indicating that MukBEF positions ori rather than vice versa.
In recent work (Hofmann et al., 2018), we found new experimental evidence that MukBEF foci position ori. By comparing the measured distribution of the MukBEF-ori separation distance to the distribution predicted if they were positioned independently, we determined that MukBEF foci and ori are not positioned to mid-cell independently of one another. Given that we know that MukBEF positioning is not strictly dependent on ori positioning, this suggests that MukBEF foci position ori. If this is the case, we reasoned that we should be able to detect this in the biased movement of ori. We measured the velocity of ori as a function of its position relative to both mid-cell and to the MukBEF focus (panel b). We found that ori displays a stronger restoring velocity (“attraction”) towards the MukBEF focus than towards mid-cell i.e. the ori is not attracted to mid-cell per se, rather it is attracted to the MukBEF focus which happens to be at mid-cell. Combined with previous results, these data strongly indicate that MukBEF foci position ori.
We then built on our previous model of MukBEF self-positioning to develop a model for the segregation and positioning of chromosomal origins. Based on our experimental results, we assumed that ori is attracted up the MukBEF gradient. Adding this assumption to our model, we found that it could indeed explain the observed ori dynamics in short cells (with a single ori and a single MukBEF focus) and showed excellent quantitative agreement with the experimental data.
However, the movement of ori up a protein gradient is not sufficient for segregation. Each ori must be partitioned to a different quarter-position. We hypothesised that feedback from ori to the MukBEF gradient can play a role. Thus, we returned to the question of the nature of the interaction between MukBEF and ori. In Bacillus subtilis the association between SMC and ori is mediated by the protein ParB, which binds to parS sites within the origin region, where it loads SMC onto the chromosome. We therefore hypothesised that MukBEF is also preferentially loaded onto the chromosome within the ori region potentially by a factor yet to be discovered. Surprisingly, we found that increased loading of MukBEF at the ori leads to an effective repulsion between ori ensuring that they are accurately partitioned (panel c). This highly non-trivial result is due to the mutual repulsion of MukBEF clusters combined with their attraction towards preferential loading sites. Furthermore, the model reproduced the observed ori-MukBEF separation distribution only in the presence of increased MukBEF loading at ori.
Hofmann*, Mäkelä*, Sherratt, Heermann and Murray† (2019) eLife 8:e46564, https://doi.org/10.7554/eLife.46564
Self-organisation of the dynamic protein complex MukBEF
The subcellular positioning of proteins is crucial for many cellular processes. This is often due to passive recruitment mechanisms. On the other hand, active mechanisms have been well-studied in the context of plasmid segregation in bacteria. Active positioning mechanisms are also believed to be responsible for the positioning of the future division site and several cytoplasmic protein clusters. Unlike plasmids however, proteins can be dynamic with rapid turnover. How such dynamic proteins can be organised and positioned was the focus of this work.

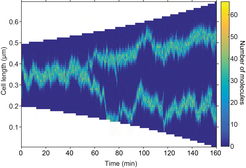
Stochastic simulation showing mid- and quarter-cell positioning. The focus splits as a result of growth. Any mis-positioning is rapidly corrected.
We investigated these questions using mathematical modelling and fluorescence microscopy taking the SMC (Structural Maintenance of Chromosomes) complex of Escherichia coli (MukBEF) as a model system. SMC complexes are ubiquitous not just in bacteria but also eukaryotes and are essential for proper chromosome segregation and condensation. It has been observed in several bacteria that fluorescent SMC fusions form a discrete number of fluorescent foci per cell. In both E. coli and Bacillus subtilis, SMC has usually either a single focus localized near mid-cell or two foci localized at approximately quarter positions. However, how these dynamic clusters are positioned has, hitherto, remained unclear. It was similarly not known what causes the length dependent transition from one to two foci.
We developed a mathematical model for the organisation of these dynamic protein clusters in which the properties of the proteins themselves are sufficient to result in self-organisation and regular positioning (Murray & Sourjik, 2017). Mathematically, the model is based on Turing pattern formation. This mechanism is widely applied to multi-cellular developmental biology but very seldom to the intracellular environment of a single cell. This is largely due to the fact that the phase of the pattern produced is not fixed i.e. different initial perturbation can give rise to patterns that are off-set from one another. We overcome this problem by the addition of a flux-balance mechanism that, in combination with stochastic effects, fixes the phase of the Turing pattern so that not only does a pattern form spontaneously due to the dynamics of the system but it is also correctly and dynamically positioned. In essence, the proteins can sense the geometry of the nucleoid and position themselves appropriately. Furthermore, the model predicts that clusters can split naturally as a result of growth.
As there are many examples of phase-fixed protein patterns especially in multi-cellular systems, the principles uncovered are likely to be applicable elsewhere. This is particularly true in developmental systems in which there is no evidence that boundary cells can physically sense their position but yet the system exhibits pattern with only internal peaks.
Murray† and Sourjik (2017) Nature Physics, 13, 1006-1013, https://doi.org/10.1038/nphys4155
Polarity switching in Myxococcus xanthus

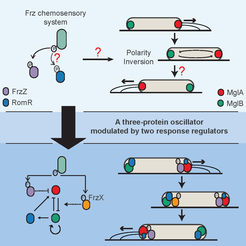
The swarming rod-shaped bacterium, Myxococcus xanthus, is a model organism for multicellular development. Its direction of movement is determined by the cell’s polarity, which is specified by the localisation of two proteins, a G-protein and its cognate GTPase-activating protein, one at each pole. However, cells are capable of very rapidly switching polarity and hence their direction of movement, with the two polar complexes rapidly switching poles. This is known to be controlled by the Frz chemosensory-like system.
In a collaboration with the lab of Tâm Mignot (CNRS, Marseille), we used mathematical modelling, genetics, and imaging to determine the mechanism behind this control (Guzzo et al. , 2018). While an initial mathematical model was insufficient to fully explain the experimental data, it lead to the discovery of the missing primary Frz output, FrzX, and a revised model of the system as a gated relaxation oscillator. The model is non-trivially consistent with all the available data and made successful predictions. Interestingly, this type of regulatory topology has, to our knowledge, not previously been found in biology. As this system involves a Ras-like GTPase, a family highly conserved throughout all eukaryotes, we expect our results to have application beyond bacteria.
Guzzo*, Murray*, Martineau*, Lhospice*, et al. (2018) Nature Microbiology, 3, 948-959, https://doi.org/10.1038/s41564-018-0203-x