Mechanism of Enzymes from Anaerobic Bacteria
Electron bifurcation and amino acid fermentations at the origin of life
Heterotrophic or autotrophic origin of life?
In microbiology bacteria are classified by the kind of their nutrition. A heterotrophic organism thrives from organic compounds like humans, whereas an autotroph obtains the whole energy and cellular material from CO2 and inorganic reductants. Currently a heterotrophic theory competes with an autotrophic theory to explain the origin of life. The idea of a heterotrophic origin of life dates back to Charles Darwin who supposed that the first organisms emerged from a “warm little pond”. Alexander I. Oparin and John B. S. Haldane formed this idea into a scientific theory that apparently was supported by the famous experiment of Stanley L. Miller and Harold C. Urey in 1953. In this experiment, lightening in an assumed reducing atmosphere generated mainly tar and hundreds of different compounds, of which only a few occur in extant life. There was no idea how the produced amino acids and other compounds assembled into a living cell. However, life is an exergonic process that continuously requires energy. Günter Wächtershäuser was the first to postulate a driving force for the abiotic synthesis of cellular material, the exergonic conversion of iron sulfide and hydrogen sulfide to pyrite and hydrogen “in statu nascendi” that – as he proposed – reduces CO2 to organic compounds via the reverse Krebs cycle. Recent geochemical research revealed that the early atmosphere contained only nitrogen and CO2. Alkaline hydrothermal vents like Lost City produce hydrogen by reduction of water with iron(II) minerals, a process called serpentinisation. Hydrogen and CO2 are the substrates of modern acetogenic bacteria and methanogenic archaea. Most importantly, both types of autotrophic organisms do not only synthesize their cell material from hydrogen and CO2 but also obtain the energy for life from this process (equation 1). Hence the autotrophic theory of the origin of life appears to be more plausible and gains an increasing number of proponents (Schönheit et al. 2016).
2 CO2 + 4 H2 ® Acetate− + H+ + 2 H2O; DG°' = −95 kJ mol−1 (equation 1).
Model organisms for the autotrophic origin use electron bifurcation
In both, acetogenic bacteria and methanogenic archaea, the electrons to reduce CO2 stem from hydrogen. Acetogens reduce one CO2 with hydrogen to formate, which is further reduced by NADH to the methyl group of acetate. The reduction of the second CO2 to CO requires a low potential ferredoxin that is obtained by “electron bifurcation”. In this system the two electrons of hydrogen are split; one electron goes to ferredoxin to become a powerful reductant, the other to NAD+. Repetition of this process affords two reduced ferredoxins and NADH. In the final step, CO combines with the methyl group (bound to tetrahydrofolate) and CoA to acetyl-CoA. Substrate level phosphorylation produces the end product acetate and ATP that is used for the activation of formate to formyl-tetrahydrofolate. The concomitant reduction of NAD+ by ferredoxin drives Rnf that generates an electrochemical Na+ gradient for additional ATP-synthesis (Buckel and Thauer, 2013).
Methanogenic archaea use almost the same acetogenic pathway but only for biosynthetic purposes. Since the energy conserving pathway from CO2 to CH4 involves no substrate level phosphorylation, CO2 is reduced by a low potential ferredoxin in one step directly to formyl-methanofuran. Further reduction by hydrogen affords methyl-methanopterin, from which the methyl group is transferred to HS-CoM, an exergonic process that generates an electrochemical Na+-gradient for ATP synthesis. Combination of methyl-S-CoM with CoB-SH leads to CoB-S-S-CoM, the heterodisulfide, and CH4. Again electron bifurcation from hydrogen to the high potential heterodisulfide enables the reduction of the low potential ‘energy rich’ ferredoxin necessary for the first step (Buckel and Thauer, 2013).
This overview of the two autotrophic pathways underlines the importance of electron bifurcation for the reduction of CO2 and hence most likely for the origin of life. Electron bifurcation has been established by Peter Mitchell already in 1976 for the Q-cycles in the membrane bound complex III (cytochrome bc1) of the respiratory chain and cytochrome b6f in photosynthesis. In 2008 the first soluble electron bifurcating system has been discovered in anaerobic butyrate forming clostridia. To date nine electron bifurcating systems are known which just represent the tip of an iceberg. Because all the new systems contain flavins, which are proposed to act as bifurcating agents rather than quinones, this process has been termed flavin based electron bifurcation (FBEB) (Herrmann et al. 2008; Li et al. 2008; Seedorf et al. 2008; Buckel and Thauer, 2013).
On the origin of heterotrophy
If the first cells were autotrophs, how did the first heterotrophs arise, and what was their substrate? We propose that autotrophs with a cell mass roughly similar to the composition of Escherichia coli was the substrate for the first chemoorganoheterotrophs. The dry mass of E. coli, a typical microbial cell, consists of about 55% protein, 25% nucleic acids, 9% lipids, 6% cell wall, 2.5% glycogen and 3% metabolites. Amino acid and nucleobase fermentations, pathways typical of anaerobic clostridia, might have been the first forms of heterotrophic carbon and energy metabolism. Ribose was probably the first abundant sugar, and the archaeal type III RubisCO pathway of nucleoside monophosphate conversion to 3-phosphoglycerate might be a relic of ancient heterotrophy. Participation of chemiosmotic coupling and flavin-based electron bifurcation in clostridial amino acid fermentations is consistent with an autotrophic origin of both metabolism and heterotrophy, as is the involvement of elemental sulfur as an electron acceptor in the facilitated fermentations of anaerobic heterotrophic archaea (Schönheit et al. 2016).
Amino acid fermenting clostridia as models for the first heterotrophic bacteria
![Fig. 1 / Activator of 2-hydroxyacyl-CoA dehydratases, also called Archerase. The central architecture of the homodimeric activator, the [4Fe-4S] cluster and helix 5 from each subunit are superimposed on the archer, King Ashurbanibal from Nineveh 650 BC, to demonstrate that the electron is injected into the dehydratase like the arrow, both powered by ATP hydrolysis.](/304491/original-1518444194.webp?t=eyJ3aWR0aCI6MTY5NiwiZmlsZV9leHRlbnNpb24iOiJ3ZWJwIiwib2JqX2lkIjozMDQ0OTF9--2c31dfcb7fe45fdfa74e455187ff0a387444106d "Fig. 1 / Activator of 2-hydroxyacyl-CoA dehydratases, also called Archerase. The central architecture of the homodimeric activator, the [4Fe-4S] cluster and helix 5 from each subunit are superimposed on the archer, King Ashurbanibal from Nineveh 650 BC, to demonstrate that the electron is injected into the dehydratase like the arrow, both powered by ATP hydrolysis.")
![Fig. 1 / Activator of 2-hydroxyacyl-CoA dehydratases, also called Archerase. The central architecture of the homodimeric activator, the [4Fe-4S] cluster and helix 5 from each subunit are superimposed on the archer, King Ashurbanibal from Nineveh 650 BC, to demonstrate that the electron is injected into the dehydratase like the arrow, both powered by ATP hydrolysis.](/304491/original-1518444194.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjMwNDQ5MX0%3D--46958dc45b307937d704876a8ea21d521590940e)
Amino acid fermentations are redox reactions. Archaea oxidize amino acids with sulfur or sulfate to acetate, CO2 and ammonia, similar to sulfate reducing bacteria. Clostridia oxidize amino acids with another amino acid or the same as electron acceptor, which has been known as Stickland reaction since the 1930s. The oxidative branches of these pathways are virtually identical to those in most organisms including humans, whereas in the reductions of amino acids are catalyzed by unusual enzymes. The apparently simplest method is the direct substitution of the α-amino group by a hydride as exemplified by the reduction of glycine to acetate and proline to 5-aminovalerate. Both reductases contain selenocysteine, but their exact mechanism remains to be elucidated; whether radicals are involved is not known yet. The most common way to reduce amino acids is the deamination to unsaturated acids that act as electron acceptors. Only aspartate (and asparagine) can eliminate ammonia directly, because they are also β-amino acids. The amino groups of lysine (2,6-diaminohexanoate) and ornithine (derived from arginine) are shifted to positions from which they are easier eliminated, catalyzed by radical S-adenosylmethionine (SAM) or coenzyme B12-dependent enzymes. The carbon skeleton of glutamate rearranges to 3-methylaspartate, also in a coenzyme B12-dependent manner (Buckel 2001b; Buckel 2005).
A more general strategy of the clostridia, by which 12 of the 20 proteinogenic amino acids are reduced to fatty acids, is the conversion of the (S)-α-amino acid by oxidation or transamination to the 2-oxoacid and reduction to the (R)-2-hydroxy acid followed by CoA-transfer. The formed (R)-2-hydroxyacyl-CoA is dehydrated to (E)-enoyl-CoA obeying a syn-geometry. This dehydration is unusual because a hydroxyl group has to be removed from the unfavorable α-position and the 3Si proton to be abstracted exhibits a pK ~ 40. The solution to this chemical problem is a radical mechanism by which a highly energized electron is injected into the dehydratase from a [4Fe-4S] cluster-containing activator (“archerase”) by hydrolysis of two ATP (Fig. 1). The heterodimeric dehydratase itself also contains two [4Fe-4S] clusters, one in each subunit; one severs as reservoir of that electron and the other as active site. As soon as the substrate binds to this cluster, it receives this electron whereby the thioester carbonyl becomes reduced to a nucleophilic ketyl radical anion, actually an ‘Umpolung’ of the electrophilic thioester. The ketyl expels the adjacent hydroxyl, whereby the pK of the β-hydrogen is reduced by 26 units. The now facile deprotonation affords an enoxy radical that is oxidized to the product enoyl-CoA and the energized electron is recycled for the next turnover. The product enoyl-CoA is either reduced by NADH to the corresponding acyl-CoA and the fatty acid is liberated by CoA-transfer. Alternatively, the CoA-transfer occurs first and is followed by reduction of the enoate, as is the case for the arylacrylates derived from the aromatic amino acids phenylalanine, tyrosine and tryptophan in Clostridium sporogenes (Buckel et al. 2012).
A special case is the fermentation of glutamate to ammonia, acetate, butyrate, CO2 and hydrogen that uses three entirely different pathways, all of which involve radicals. In these pathways glutamate is converted first to fermentable intermediates, either to crotonyl-CoA or to pyruvate. The conversion to crotonyl-CoA in Acidaminococcus fermentans follows the pathway via the (R)-2-hydroxy acids to glutaconyl-CoA, an enoyl-CoA. A biotin-containing membrane enzyme catalyzes the decarboxylation of glutaconyl-CoA to crotonyl-CoA, whereby the free energy generates an electrochemical Na+-gradient for glutamate import and ATP-synthesis (Buckel 2001a; Wendt et al. 2003). Crotonyl-CoA disproportionates to acetate, butyrate and hydrogen. The path to pyruvate in Clostridium tetanomorphum starts with a coenzyme B12-induced radical rearrangement of (S)-glutamate to (2S,3S)-3-methylaspartate, which allows the facile elimination of ammonia to mesaconate (methylfumarate). Hydration affords (S)-citramalate that is cleaved to acetate and pyruvate. In the following steps, ferredoxin oxidizes pyruvate to CO2 and acetyl-CoA, from which butyrate is synthesized (Buckel 2001b).
The third path of glutamate fermentation involves two organisms. At low pH Escherichia coli and other bacteria decarboxylate glutamate to 4-aminobutyrate (γ-aminobutyrate, GABA), whereby one proton is consumed. Clostridium aminobutyricum ferments 4-aminobutyrate via crotonyl-CoA to acetate, butyrate and ammonia. The key enzyme of this pathway is 4-hydroxybutyryl-CoA dehydratase (AbfD) that catalyzes the reversible dehydration of 4-hydroxybutyryl-CoA to crotonyl-CoA via radical intermediates. Recently, this radical enzyme also has been identified in a new CO2 fixation pathway in Archaea (Berg et al. 2007). The [4Fe-4S] cluster and FAD-containing AbfD, whose crystal structure has been solved (Martins et al. 2004), catalyzes an oxygen sensitive and chemical difficult dehydration, because the non-activated 3Si-proton of 4-hydroxybutyryl-CoA has to be removed (pK ~ 40). The successful production of variants of recombinant AbfD in E. coli demonstrated that all highly conserved amino acids around the active center are essential for activity. The low residual activity of the Y296F variant (<1%) suggested that a tyrosine radical may be involved in catalysis. Probably this radical forms already upon substrate binding, prior to the actual dehydration, because crotonyl-CoA as well as its analogue butyryl-CoA and even free CoA induce a partial one-electron reduction of the [4Fe-4S]2+ cluster and FAD, whereby Y296 assisted by E455 as base might act as donor. The dyad of T190 and E257, which is in hydrogen bonding distance to N5 of FAD, reversibly protonates the thereby formed semiquinone anion to FADH• (Zhang et al. 2015). The dehydration proceeds with anti-elimination of the 2Re-hydrogen and the 3Si-hydrogen and with retention of the configuration at C4 during substitution of the hydroxyl group by hydrogen (Friedrich et al. 2008). We think that histidine 292 removes the 2Re proton to give the enolate. In contrast to the dehydration of 2-hydroxyacyl-CoA, we propose a one-electron oxidation of the enolate of 4-hydroxybutyryl-CoA by FADH• to an enoxy radical, which lowers the pK of the 3Si proton from about 40 to 14 (Smith et al. 2003). The dyad deprotonates the enoxy radical to an allylic ketyl radical anion that expels the hydroxyl group bound to Fea of the cluster. The formed FADH– reduces the dienoxy radical to the dienolate that is protonated by E455 to the product (Zhang et al. 2015).
Reduction of ferredoxin or flavodoxin coupled to butyrate formation
The fermentation of sugars and amino acids to butyrate is a characteristic feature of anaerobic bacteria of the Firmicutes phylum. In the human gut, butyrate is derived from the microbial degradation of fibers, i.e. oligosaccharides or peptides which are non-digestible by human enzymes. Butyrate nourishes the epithelia cells of the gut and thus is essential for human health. Until recently the production of butyrate by bacteria was regarded as a mere disposal for reducing equivalents as was the reduction of the enoyl-CoAs and unsaturated fatty acids formed in the reductive branches of the amino acid fermentations. The discovery of electron bifurcation has established that the butyrate forming pathway contributes to hydrogen formation and energy conservation, because the exergonic reduction of crotonyl-CoA to butyryl-CoA by NADH is tightly coupled to the endergonic reduction of ferredoxin (Fd−) also by NADH. This process has been called electron bifurcation, because the two electrons supplied by NADH (E°' = –320 mV) move to different potentials: one electron goes to the high potential crotonyl-CoA (E°' = –10 mV) and the other to the low potential ferredoxin (E°' = –405 mV; E' ≤ –500 mV). Repetition of this process affords butyryl-CoA and two reduced ferredoxins (Fd2−). This “energy rich” electron carrier gives rise to molecular hydrogen and/or recycles NADH, mediated by a ferredoxin-NAD+ reductase also called Rnf. The energy difference between reduced ferredoxin and NAD+ of ca. 200 mV is used by this integral membrane enzyme to generate an electrochemical Na+-gradient (Chowdhury et al. 2014, 2015, 2016; Buckel 2014).
 and butyryl-CoA dehydrogenase (Bcd) catalyzes the concomitant reduction of crotonyl-CoA and two ferredoxin by two NADH. One NADH is regenerated by reduction of NAD by ferredoxin catalyzed by Rnf (RF = riboflavin). The free energy of this reaction pumps Na+ out of the cell. The formed electrochemical Na+ gradient drives the synthesis of ATP.")
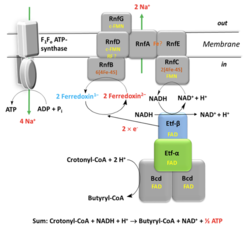
Fig. 2 / Energy conservation in amino acid fermenting anaerobes, e.g. Acidaminococcus fermentans. The electron bifurcating complex composed of electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) catalyzes the concomitant reduction of crotonyl-CoA and two ferredoxin by two NADH. One NADH is regenerated by reduction of NAD by ferredoxin catalyzed by Rnf (RF = riboflavin). The free energy of this reaction pumps Na+ out of the cell. The formed electrochemical Na+ gradient drives the synthesis of ATP.
In A. fermentans the electron bifurcating reduction of crotonyl-CoA is catalyzed by two enzymes. One is a heterodimeric electron transferring flavoprotein (EtfAB), which contains one FAD in each subunit (α and β-FAD on subunit A and B, respectively), and the other a homotetrameric butyryl-CoA dehydrogenase (Bcd) with one FAD (d-FAD) in each subunit. Based on protein crystallography and enzymatic assays, we propose that NADH reduces b-FAD to the hydroquinone (b-FADH−), which delivers one electron further to the high potential α-FAD that is 14 Å apart to become a stable anionic semiquinone (α-FAD·−). The remaining low potential and highly reactive b-FADH· donates its electron further to ferredoxin. Then α-FAD·−, located on a flexible domain, swings to the FAD of Bcd (d-FAD) and transfers its electron to yield d-FADH·. Finally, the second round generates d-FADH− that reduces crotonyl-CoA to butyryl-CoA (Chowdhury et al. 2014).
By growing A. fermentans on a medium with low iron, ferredoxin is replaced by flavodoxin that contains riboflavin-5'-phosphate (FMN) as prosthetic group. Due to the different colors of the three redox states, yellow quinone (FMN), blue semiquinone (FMNH•) and colorless hydroquinone (FMNH–), the redox reactions can be easily followed. Whereas the redox potential of FMN/FMNH• ( E°' = –60 mV) is surprisingly high, that of FMNH•/FMNH– ( E°' = –430 mV) is similar to ferredoxin. Incubation with crotonyl-CoA, NADH in the presence of Etf and butyryl-CoA dehydrogenase leads to the reduction of flavodoxin. As soon as the FMN of flavodoxin is completely converted to the blue FMNH•, a faster reduction rate affords almost quantitatively FMNH–. Hence the effective redox potential of the FMNH•/FMNH– couple approaches ≤ –500 mV. The hydroquinone can be reoxidized to the semiquinone either by protons catalyzed by a hydrogenase or by NAD+ mediated by membranes of A. fermentans containing high levels of Rnf. The latter reaction requires Na+ or Li+ ions for activity (Chowdhury et al. 2016) (Fig. 2).
Over 50 years ago, Baldwin and Milligan reported that Etf and butyryl-CoA dehydrogenase of the anaerobic rumen bacterium Megasphaera elsdenii catalyzed the reduction of crotonyl-CoA to butyryl-CoA by NADH but ferredoxin was not required. We demonstrated that under anaerobic conditions Etf + Bcd from M. elsdenii bifurcated as efficient as Etf + Bcd from A. fermentans. Under aerobic conditions used by Baldwin and Milligan and in the presence of catalytic amounts of crotonyl-CoA or butyryl-CoA, however, Etf + Bcd acted as NADH oxidase producing superoxide and H2O2, for which ferredoxin was not required. We hypothesize that during bifurcation oxygen replaces ferredoxin to be reduced to superoxide. In addition the formed butyryl-CoA is re-oxidized by a second oxygen molecule to crotonyl-CoA resulting in an overall stoichiometry of 2 NADH consumed and 2 H2O2 formed. Due to the production of reactive oxygen species (ROS), electron bifurcation can be regarded as the Achilles’ heel of anaerobes when exposed to air (Chowdhury et al. 2015).
The great oxygenation event
This review gives an idea how most organisms thrived before the great oxygenation event (GOE), which was caused by the action of photosynthetic cyanobacteria about 2.3 billion years ago. Due to the emergence of oxygen, all reactions with lower standard redox potentials than that of NAD+/NADH (E°' = –320 mV) disappeared in aerobes. Thus all bifurcating systems and other reactions with ferredoxin or flavodoxin as electron acceptors or donors were abandoned. Furthermore, oxygen allowed an almost 6-fold expansion of the length of the respiratory chain from ferredoxin to NAD+ of ΔE = 200 mV to that from NADH to oxygen of ΔE = 1140 mV, which increased the amount of conserved energy by this factor. Notably also the extremely oxygen sensitive 2-hydroxacyl-CoA dehydratases are absent in aerobes due to the energized electron (E' ~ –600 mV) required for catalysis. In contrast the clostridial 4-hydroxybutyryl-CoA dehydratases survived in aerobic CO2 fixing archaea. Although these enzymes are proposed to use also ketyl radicals as intermediates, the archaeal type is about 100-times less oxygen sensitive than the clostridial version (Berg et al. 2007). Hence evolution has managed to protect many radical enzymes from oxygen, but the electron donors with redox potentials of <–320 mV disappeared because they rapidly reduce oxygen to the ‘dangerous’ superoxide (O2/O2•–, E°' = –330 mV) also called reactive oxygen species (ROS) (Chowdhury et al. 2015).